Nephroquizz : Le CJN vous met au défi !
Comme chaque année, le CJN organise la session Néphroquizz du congrès de la SFNDT. Elle aura lieu le vendredi 13 octobre à 9h en salle Gallieni ½.
Cette année, on vous laisse dormir un peu plus longtemps pour être en forme et phosphorer sur trois cas de néphrologie exceptionnels ! Les trois meilleurs auront la chance de repartir avec un ouvrage de référence en néphrologie, transplantation ou dialyse. Les 3 cas seront affichés à proximité du stand du CJN dès le début du congrès. A vos neurones !
Et pour ceux qui auraient besoin d’échauffement, on vous remet les cas de l’année dernière !

Cliquez sur les poster pour les agrandir
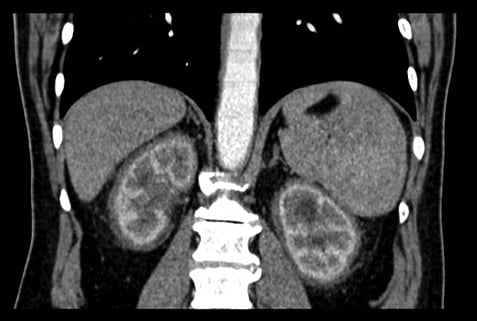
Cas n° 1 : Maladie d’Erdheim-Chester
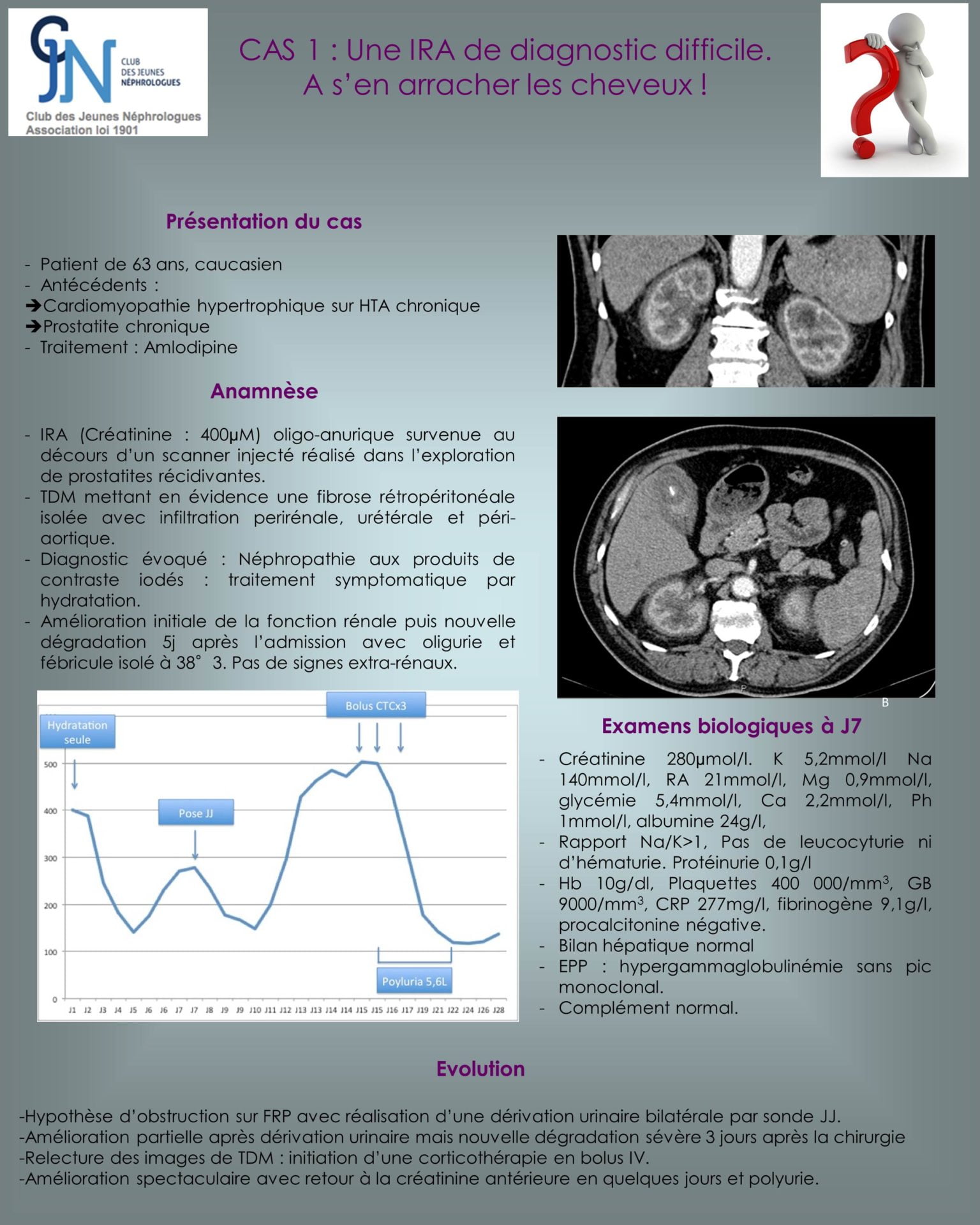
La maladie d’Erdheim-Chester est une forme d’histiocytose non langerhansienne avec atteinte multisystémique : altération de l’état général, douleurs osseuses, exophtalmie, atteinte hypophysaire (diabète insipide…), insuffisance rénale, atteinte cardiovasculaire (manchon aortique, HTA, péricardite…) et du système nerveux central (hypertension intracrânienne, paralysie des nerfs crâniens, cécité corticale…) . La maladie est rare et d’étiopathogénie inconnue).
Elle atteint principalement les hommes entre 40 et 60 ans (rares cas pédiatriques). Le signe pathognomonique est l’ostéosclérose des os long (principalement l’extrémité distale des membres inférieurs), responsables de douleurs osseuses.
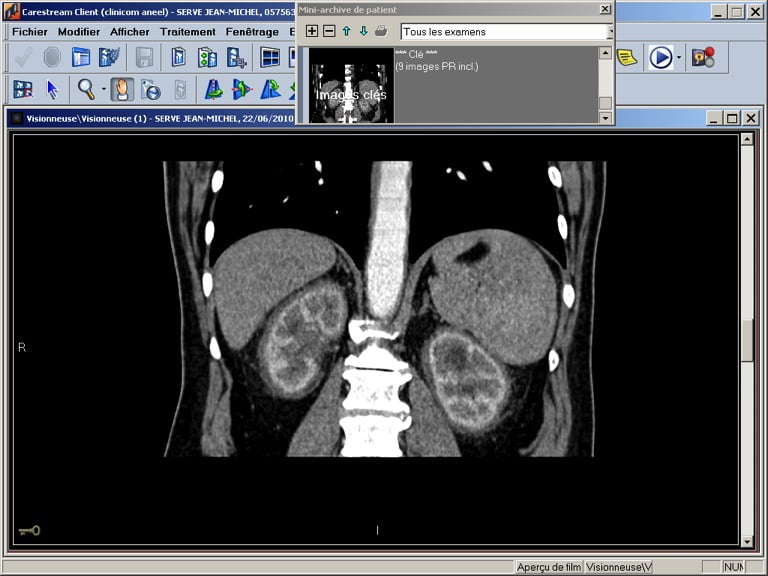
 L’atteinte rénale est à type de fibrose rétropéritonéale avec l’aspect classique de « reins chevelus » (« hairy kidneys »).
L’atteinte rénale est à type de fibrose rétropéritonéale avec l’aspect classique de « reins chevelus » (« hairy kidneys »).
L’insuffisance rénale est donc obstructive avec généralement hydronéphrose bilatérale. L’atypie dans ce cas est l’insuffisance rénale obstructive sans dilatation des cavités pyélocalicielles (du fait de l’infiltration envahissant les reins et les uretères), et ayant conduit à retarder le diagnostic étiologique.
Le diagnostic de la maladie d’Erdheim-Chester est anatomopathologique (xanthogranulomatose CD68+, CD1a-). Le traitement de première intention est l’interféron-a. Le pronostic de la pathologie est variable avec un taux de survie à 5 ans autour de 70 %. Le pronostic est moins bon en cas d’atteinte du SNC.
Référence :
Haroche J, Cohen-Aubart F, Arnaud L, Hervier B, Charlotte F, Drier A, Gorochov G, Grenier PA, Cluzel P, Maksud P, Emile JF, Amoura Z. [Erdheim-Chester disease]. Rev Med Interne. 2014 Nov;35(11):715-22.
Cas n°2 : Mutation de CYP24A1
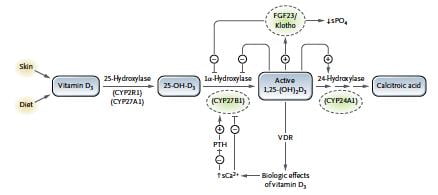
Cette pathologie a été découverte en 2011 par l’équipe de Konrad par l’exploration génétique d’enfants ayant présenté une hypercalcémie alors qualifiée d’idiopathique. CYP24A1 est l’enzyme qui dégrade la vitamine D active.
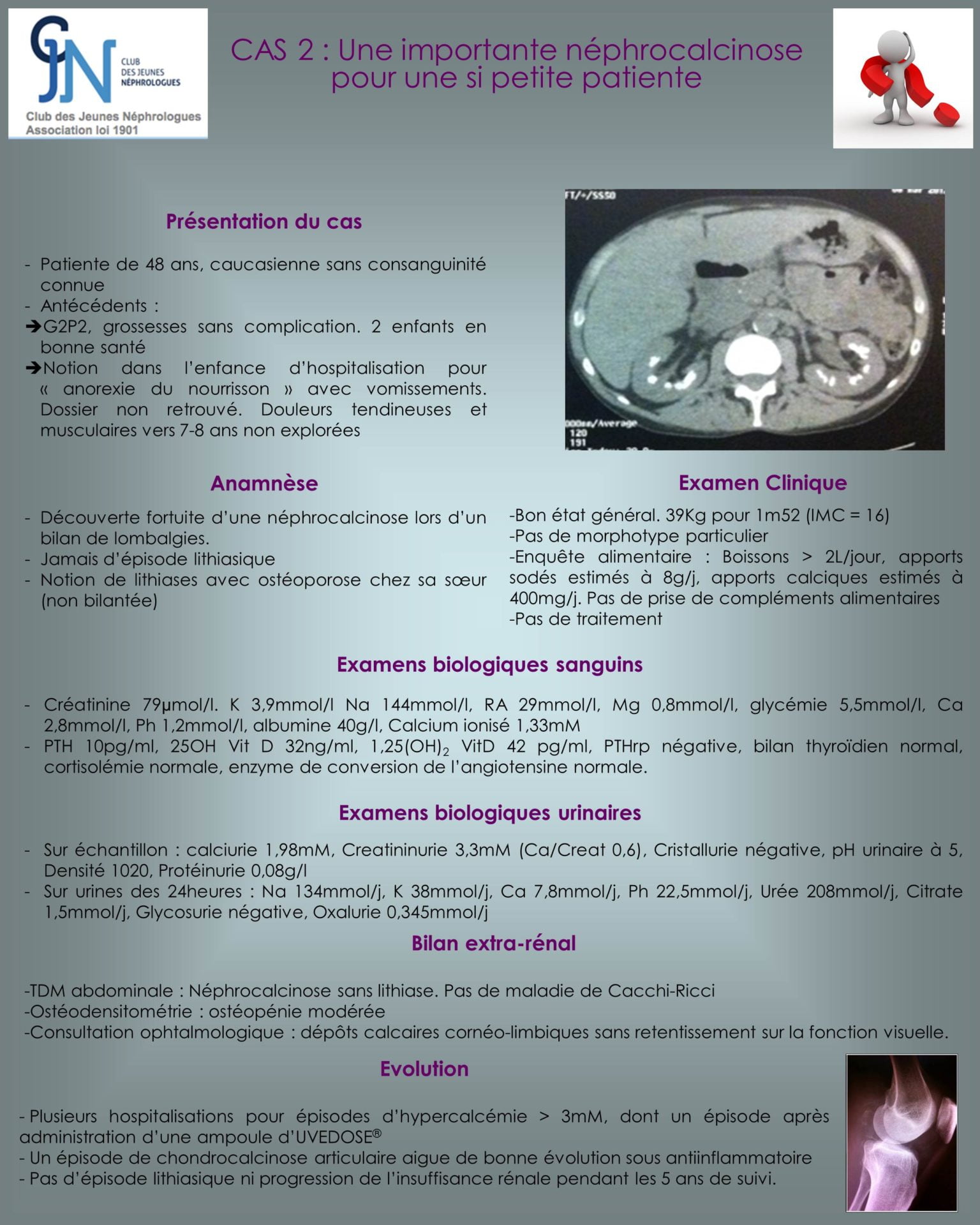
 Les patients atteints de cette pathologie ont une néphrocalcinose précoce, des lithiases calciques et des épisodes d’hypercalcémie spontanés ou provoqués par la prise de vitamine D. L’augmentation de la 1,25OH2 vitamine D est fortement évocateur mais n’est pas systématiquement retrouvé, même pendant l’épisode d’hypercalcémie. Les autres causes d’hypercalcémie seront éliminées et l’analyse génétique confirmera le diagnostic. L’atteinte des hétérozygotes est à ce jour mal évaluée. Certains présentent des lithiases calciques sans néphrocalcinose. Le traitement repose principalement sur l’arrêt d’une éventuelle supplémentation par vitamine D et la protection solaire. Du ketoconazole peut être utilisé (diminue l’activité 1-a hydroxylase des macrophages). Le pronostic est bon malgré le risque d’insuffisance rénale chronique du fait de la néphrocalcinose, et les calcification extra rénales (chrondrocalcinose, calcifications vasculaires…).
Les patients atteints de cette pathologie ont une néphrocalcinose précoce, des lithiases calciques et des épisodes d’hypercalcémie spontanés ou provoqués par la prise de vitamine D. L’augmentation de la 1,25OH2 vitamine D est fortement évocateur mais n’est pas systématiquement retrouvé, même pendant l’épisode d’hypercalcémie. Les autres causes d’hypercalcémie seront éliminées et l’analyse génétique confirmera le diagnostic. L’atteinte des hétérozygotes est à ce jour mal évaluée. Certains présentent des lithiases calciques sans néphrocalcinose. Le traitement repose principalement sur l’arrêt d’une éventuelle supplémentation par vitamine D et la protection solaire. Du ketoconazole peut être utilisé (diminue l’activité 1-a hydroxylase des macrophages). Le pronostic est bon malgré le risque d’insuffisance rénale chronique du fait de la néphrocalcinose, et les calcification extra rénales (chrondrocalcinose, calcifications vasculaires…).
Référence :
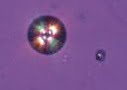
Cas n° 3 : Déficit en APRT
Les lithiases de 2,8-dihydroxyadénine sont secondaires à un déficit en adénine phosphoribosyl transférase (APRT). C’est une pathologie autosomique récessive évoquée par la survenue (souvent asymptomatique et découverte tardivement) de lithiases radiotransparentes avec cristaux d’aspect spécifique en croix de malte.
 Le diagnostic de certitude repose sur l’analyse spectrophotométrique des calculs et la cristallurie, complétée par la mesure de l’activité enzymatique de l’APRT. Les cristaux débutent dans l’enfance. L’insuffisance rénale apparaît par précipitation intratubulaire des cristaux, et est rarement complètement réversible. Le traitement associe des mesures hygiéno-diététiques (régime pauvre en purines, boissons abondantes et bien réparties sur le nycthémère) à l’allopurinol. Il n’ y a pas d’intérêt à alcaliniser les urines (la solubilité de la 2,8-dihydroxyadénine ne dépend pas du pH).
Le diagnostic de certitude repose sur l’analyse spectrophotométrique des calculs et la cristallurie, complétée par la mesure de l’activité enzymatique de l’APRT. Les cristaux débutent dans l’enfance. L’insuffisance rénale apparaît par précipitation intratubulaire des cristaux, et est rarement complètement réversible. Le traitement associe des mesures hygiéno-diététiques (régime pauvre en purines, boissons abondantes et bien réparties sur le nycthémère) à l’allopurinol. Il n’ y a pas d’intérêt à alcaliniser les urines (la solubilité de la 2,8-dihydroxyadénine ne dépend pas du pH).
Référence :
Bouzidi H, Lacour B, Daudon M. [2,8-dihydroxyadenine nephrolithiasis: from diagnosis to therapy]. Ann Biol Clin (Paris). 2007 Nov-Dec;65(6):585-92. Review
