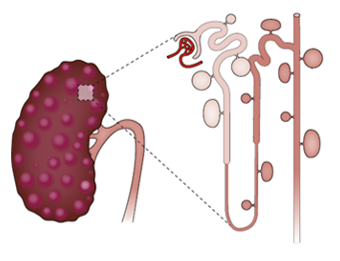
Les kystes apparaissent dès la vie utérine et croissent durant toute la vie de l’individu
Dans la PKAD , ils peuvent se développer à partir de toutes les régions du rein mais surtout au niveau des parties distales, principalement le canal collecteur.
PKD1 et PKD2 codent respectivement pour la polycystine 1 et 2.
Les polycystines sont localisées dans plusieurs compartiments de la cellule épithéliale tubulaire : cil primaire, jonction cellulaire, réticulum endoplasmique pour PC2. C’est la perte de fonction des polycystines qui aboutit à la croissance des kystes et à la fibrose rénale détruisant progressivement le parenchyme rénal.
Les polycystines ont une expression ubiquitaire expliquant « l’atteinte systémique » de la PKAD et donc les atteintes extra rénales de cette maladie : kyste hépatique, anévrysmes cérébraux, atteintes valvulaires cardiaques…
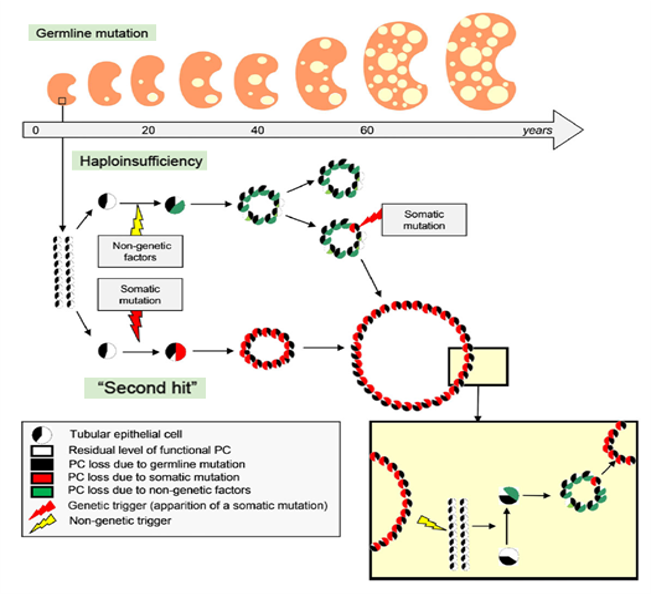
Plusieurs modifications sont nécessaires pour expliquer le phénotype: tout commence par une mutation germinale provoquant une insuffisance haplotypique, auquel s’ajoute un second hit génétique (mutation somatique, association d’autres gènes) ou non génétique (facteurs environnementaux) responsable d’une diminution de l’expression de PKD1 ou PKD2 sous les 50%. Il est nécessaire qu’un troisième hit soit présent, dans le cas le plus fréquent il s’agit d’un signal de prolifération de la cellule mutée pour les deux allèles.
Dans un contexte de mutations génétiques liées à la PKD, l’activation aberrante ou chronique de certaines voies moléculaires (voies de développement mécanismes de réparation/remodelage) peuvent entraîner une exacerbation de la maladie.
Cornec-Le Gall E et al., .Hum Mutat. 2014 Dec;35(12):1393-406