
BJN#90 – Un nouveau traitement dans la maladie de Fabry ?
Treatment of Fabry’s Disease with the Pharmacologic Chaperone Migalastat
Merci à Flora Brunner, Néphrologue à Marseille et membre du conseil scientifique du CJN, pour cette synthèse bibliographique. Vous aussi, n’hésitez pas à nous envoyer vos lectures !
Introduction
La maladie de Fabry est une maladie génétique liée à l’X causée par l’activité déficiente de l’alpha-galactosidase A lysosomale qui entraîne l’accumulation de glycosphingolipides principalement le globotriaosylcéramide (GL-3) entraînant une atteinte multi-organes et une réduction de l’espérance de vie. Le traitement actuel consiste en une enzymothérapie substitutive injectée toutes les 2 semaines. Ses inconvénients sont représentés principalement par la lourdeur du traitement (perfusions bi-mensuelles à vie) et par la formation d’anticorps avec par la suite une mauvaise tolérance des perfusions. Le migalastat, une molécule chaperon prise par voie orale, stabilise certaines formes mutées d’alpha-galactosidase, augmentant ainsi l’activité enzymatique au niveau des lysosomes.
Matériels et méthodes
Les patients éligibles avaient une maladie de Fabry confirmée génétiquement (plus ou moins avec une mutation sensible au traitement par migalastat). Ils étaient randomisés pour recevoir en phase 1 soit 6 mois de migalastat soit un placebo ; puis en phase 2 les patients des 2 groupes recevaient en ouvert 6 mois de migalastat.
L’objectif principal était le pourcentage de patients présentant une réduction de plus de 50% dans le nombre d’inclusions de GL-3 au niveau des capillaires de l’interstitium rénal à M6. Les objectifs secondaires comportaient l’évaluation du taux urinaire de GL-3, de la fonction rénale, de la protéinurie et de la tolérance du traitement.
Les objectifs tertiaires comprenaient l’évaluation de la fonction cardiaque, des signes cliniques liés à la maladie et de l’activité de l’alpha-galactosidase au niveau des globules blancs.
Résultats
Le critère de jugement principal, portant sur les patients porteurs ou non d’une forme mutée d’alpha-galactosidase sensible au traitement par migalastat, n’a pas montré de différence entre les 2 groupes : 13 des 32 patients (41%) recevant le migalastat versus 9 des 32 patients (28%) recevant le placebo avaient répondu à 6 mois (p=0.3).
Dans une analyse post-hoc pré-spécifiée qui a analysé 45 patients porteurs d’une mutation sensible au traitement par migalastat, les auteurs montraient que 6 mois de traitement étaient associés à une réduction significative dans le nombre moyen d’inclusions de GL-3 par capillaire péri-tubulaire par rapport au placebo : -0.25 ± 0.10 versus 0.07 ± 0.13 ; p = 0.008 (Fig1)
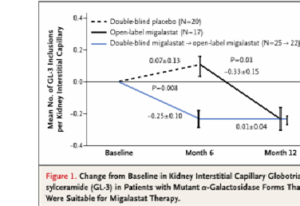
Parmi les patients porteurs d’une mutation sensible au traitement traités jusqu’à 24 mois par migalastat, les variations annualisées du DFGe et du DFGm étaient respectivement de – 0.30 ± 0.66 et – 1.51 ± 1.33 ml/min/1.73m2.
L’indice de masse du ventricule gauche diminuait significativement par rapport à la base (- 7.7g/m2 [- 15.4 ;-0.01]), surtout lorsqu’il existait une hypertrophie ventriculaire gauche (-18.6g/m2 [-38.2 ; 1]. La sévérité des symptômes digestifs (diarrhées, reflux) diminuait.
La tolérance du traitement par migalastat était bonne. Seulement 2 effets secondaires ont été considérés comme reliés au traitement par les investigateurs : asthénie et paresthésie spontanément résolutives sans nécessité d’arrêter le traitement.
Conclusion
Parmi les patients randomisés porteurs ou non d’une mutation sensible au traitement par migalastat, on ne retrouvait pas de différence significative sur la réduction du nombre d’inclusions de GL -3 au niveau des capillaires péri-tubulaires entre les 2 groupes.
Les plus du papier
- étude clinique portant sur un nouveau traitement concernant une pathologie rare mais compliquée d’une morbi-mortalité importante notamment sur le plan rénal et cardiaque
Les critiques
- changement de kit génétique en cours d’étude avec initialement des patients inclus qui étaient porteurs d’une mutation non sensible au traitement par migalastat, ce qui a pu influencer les résultats portant sur le critère de jugement principal
- traitement comparé à un placebo et non pas au traitement par enzymothérapie substitutive
